An international team of researchers has analyzed remains from ten archaeological sites in England, France, Germany, Russia, and Switzerland to gain insight into the different stages of the second plague pandemic (14th-18th centuries) and the genetic diversity of Yersinia pestis during and after the Black Death. In a study published in Nature Communications, the researchers reconstructed 34 Y. pestis genomes, tracing the genetic history of the bacterium, which revealed key insights into the initiation and progression of the second plague pandemic in Europe.
The second plague pandemic, which began with the Black Death in the mid-14th century and continued with devastating outbreaks in Europe and the vicinity until the 18th century, decimated the continent, causing the death of up to 60% of the population. But where did this strain of Yersinia pestis, the plague causing bacterium, come from? And how did it evolve and expand once it arrived?

A likely point of entry for Y. pestis during the second pandemic
[ad_336]
Despite the ubiquity of the Black Death in historical texts and the popular imagination, the entry point of the Y. pestis bacterium at this time and the route it traveled through Europe remain unclear, due to a lack of data from early outbreaks and a general scarcity of published ancient Y. pestis genomes. In the current study, researchers reconstructed plague genomes from the teeth of 34 individuals, including two from Laishevo, in the Volga region of Russia, and found a single strain that is ancestral to all second pandemic strains. In addition, the researchers observe an absence of genomic diversity from samples during the Black Death.
“These findings indicate a single entry of Y. pestis into Europe through the east”, explains first author Maria Spyrou of the Max Planck Institute for the Science of Human History. “However, it is possible that additional interpretations may be revealed with future discoveries of un-sampled diversity in western Eurasia”.

Persistence of Y. pestis within Europe
Although the researchers found that the European-wide Black Death was likely caused by a single strain, analysis of genomes from later in the pandemic shows the emergence of a lineage displaying a higher genetic diversity. “In the later phase of the second pandemic, we see the development of multiple branches within Europe, which suggests that plague was maintained in different local foci”, says Marcel Keller, co-first author of the Max Planck Institute for the Science of Human History.
“No modern descendants of this lineage have been found to date, possibly indicating the extinction of these reservoirs.”
[rand_post]
The researchers also identified a deletion including two virulence-related genes from genomes within this second lineage. Interestingly, genomes from the late stages of the first plague pandemic have shown a deletion in the same region.
“Given that this deletion occurred in lineages from the first and second pandemic, both now extinct, determining how these genes impact maintenance in human and flea hosts would be an important area for future study”, comments Kirsten Bos, research group leader of the Max Planck Institute for the Science of Human History.
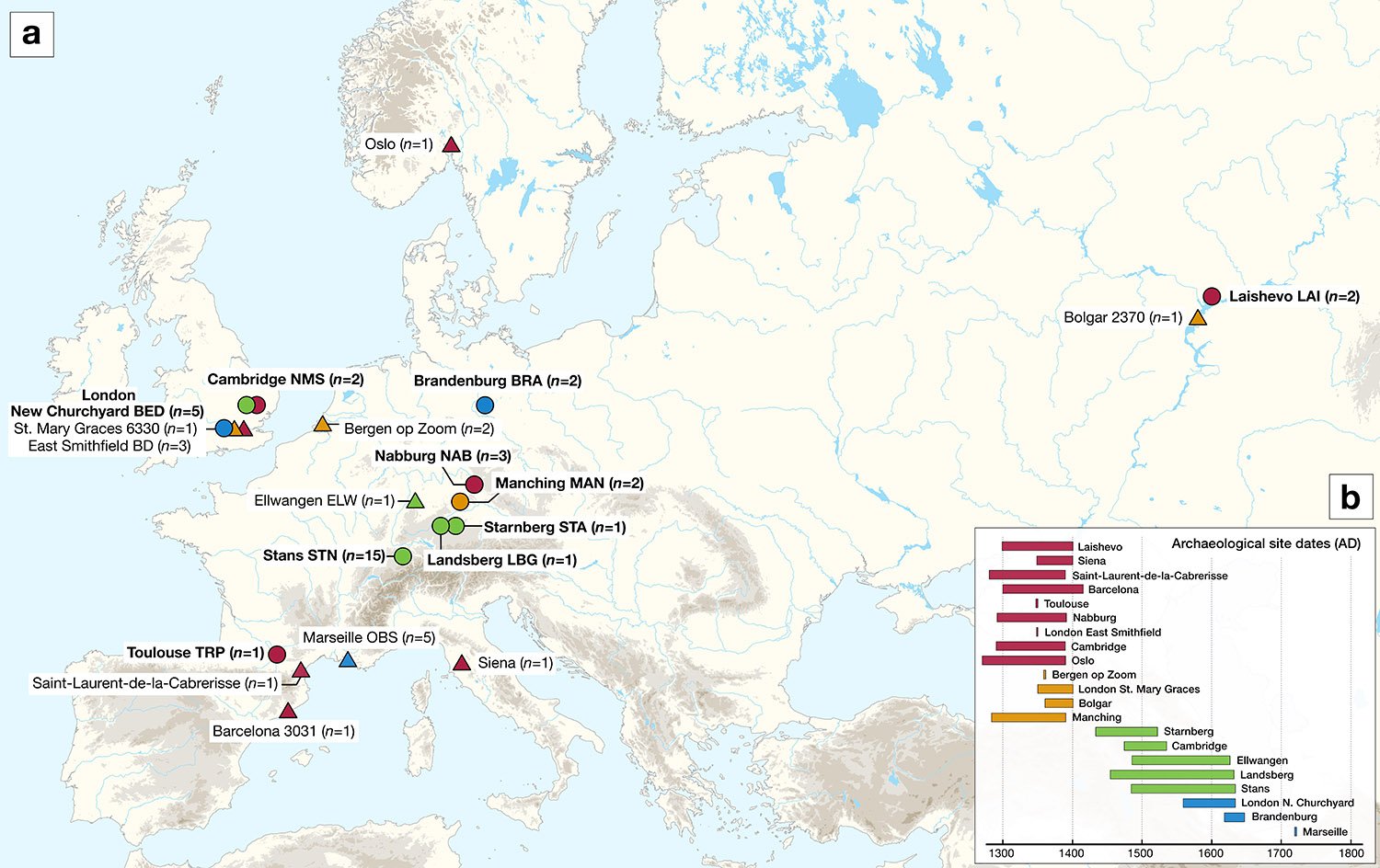
The current study provides new perspectives into the initiation and progression of the second plague pandemic and adds significantly to the database of published ancient Y. pestis genomes.
“We have shown that extensive analysis of ancient Y. pestis genomes can provide unique insights into the microevolution of a pathogen over a period of several hundred years”, says senior author Johannes Krause, Director of the Department of Archaeogenetics at the Max Planck Institute for the Science of Human History.
In the future, integrating this data into disease modelling efforts, in conjunction with data from other areas such as climate science, epidemiology and history, will be important for better understanding the second plague pandemic.